
Amyotrophic lateral sclerosis (ALS) is commonly referred to as Lou Gehrig’s disease, after the famous baseball player who was diagnosed with it.
It is a chronic, progressive neurodegenerative disease that affects nerve cells in the brain and spinal cord.
ALS causes muscle weakness, muscle cramps, atrophy, and paralysis, leading to respiratory failure and death. ALS is a rare disease, affecting about 1 in 10,000 people worldwide, and most commonly diagnosed between the ages of 40 and 70.
Motor neurons run from the brain to the spinal cord, and from the spinal cord to muscles throughout the body. In ALS, these motor neurons deteriorate and affect an individual’s ability to initiate and control muscle movement.
Over time, the brain loses this ability entirely. Individuals often lose voluntary muscle actions such as speaking, eating, moving, and breathing. Other examples of voluntary movements include reaching out to grab something (an action controlled by the arm), or walking (an action controlled by the legs).
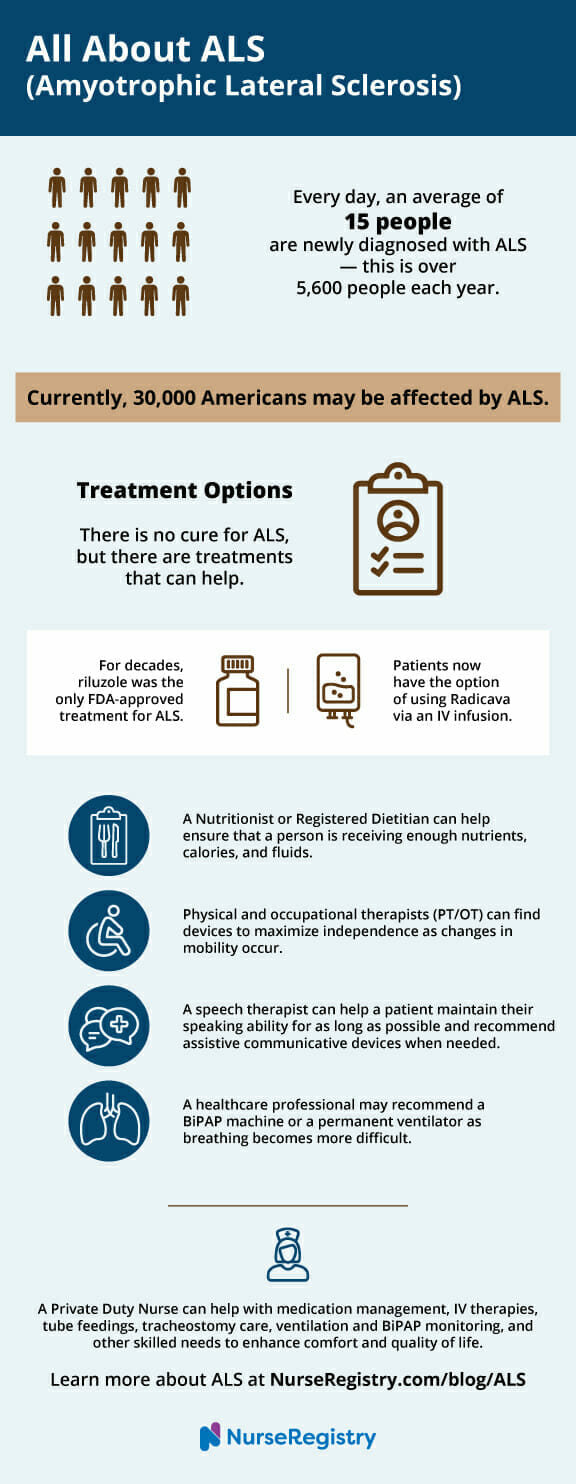
Currently, 30,000 people may be affected by ALS in the United States. On average, 5,600 people this year will be diagnosed with ALS.
The exact cause of ALS is unknown, but it is believed to be a combination of genetic and environmental factors.
Unfortunately, there is no cure for ALS, and the average life expectancy after diagnosis is 2-5 years. However, there are treatments available that can help manage symptoms and improve quality of life for those living with the disease.
Understanding ALS is crucial for early diagnosis and management of the disease. In this article, we will explore the symptoms, progression, diagnosis, treatments, and future directions of ALS, as well as answer some frequently asked questions about the disease.
In this article, you will learn about
Key Takeaways
- ALS is a progressive neurodegenerative disease that affects the nerve cells responsible for controlling voluntary muscle movement.
- The exact cause of ALS is unknown, and there is no cure for the disease.
- Early diagnosis and management of ALS is important for improving quality of life for those living with the disease.
The Biology of ALS
IN THIS ARTICLE
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease that affects the nerve cells in the brain and spinal cord. It is a type of motor neuron disease that causes the death of the nerve cells that control voluntary muscles.
As a result, the muscles gradually weaken and waste away, leading to difficulty in speaking, swallowing, and breathing.
The disease is characterized by the degeneration of two types of motor neurons: the upper motor neurons and the lower motor neurons.
The upper motor neurons are located in the brain, while the lower motor neurons are located in the spinal cord. The upper motor neurons send signals to the lower motor neurons, which in turn control the muscles in the body.
In ALS, both types of motor neurons degenerate, leading to the loss of muscle control and eventual paralysis.
The exact cause of ALS is not known, but several factors have been implicated in its development, including genetics, environmental factors, and lifestyle factors.
Mutations in certain genes, such as the SOD1 gene, have been linked to the development of ALS. Environmental factors, such as exposure to toxins, have also been implicated in the disease.
1. Types and Risk Factors
There are two forms of ALS.
Sporadic is the most common form in the US and accounts for 90 to 95% of all cases.
Familial ALS (FALS) accounts for 5 to 10 percent of cases in the US. FALS is inherited from a parent. For affected individuals, each offspring carries a 50 percent risk of inheriting the gene mutation. Both sporadic and familial ALS share the same symptoms and progression. All forms of ALS are not contagious.
Amyotrophic lateral sclerosis usually affects people between the ages of 40 and 70.
Evidence has shown that military veterans are twice as likely to be diagnosed with the disease as the general public, especially those who have served during the Persian Gulf War of 1991, though the reasons are unknown. ALS has also been shown to affect men more frequently than women; again, the reasons behind this are unknown.
2. Symptoms

The early symptoms of ALS can be subtle and easily overlooked, making the disease difficult to diagnose in its early stages.
Some of the early symptoms of ALS may include muscle weakness, muscle cramps, and twitching.
These symptoms may initially be limited to one area of the body, such as the hands or feet, but will eventually spread to other areas.
As voluntary muscle functions are affected, individuals with early-stage ALS may notice symptoms such as:
- Twitching (fasciculations) and cramping of muscles
- Occasional jerking of the arms or legs
- Stiffness or spasticity
- Loss of motor controls in the hands and arms, which results in difficulty unlocking doors, writing, or other fine motor tasks
- Tripping or stumbling (weakness in the legs)
- Difficulty speaking or swallowing
- Slurred or thick, nasally speech
- Difficulty projecting the voice
- Generalized fatigue
ALS may be present before symptoms become noticeable. This occurs when remaining functioning nerve cells compensate for lost or damaged nerve cells, which may result in generalized fatigue.
As muscle cells deteriorate and the disease progresses, patients may experience:
- Difficulty breathing
- Difficulty with speech and communication (slurred speech)
- Paralysis
Symptoms often begin in the extremities, and move towards the center of the body. Typically, one side of the body will be more affected than the other. As the disease progresses, complete paralysis may occur, except for the muscles of the eyes.
Rate of Progression
The rate of progression of ALS varies from person to person and can be difficult to predict. Some people with ALS experience a slow, gradual decline in muscle function, while others experience a more rapid decline. In general, however, ALS is a progressive disease that gets worse over time.
While there is no cure for ALS, there are treatments available that can help manage the symptoms and slow the progression of the disease. Physical therapy, occupational therapy, and speech therapy can all be helpful in maintaining muscle function and improving quality of life for people with ALS.
3. Diagnosis
An ALS diagnosis is made by a neurologist with a physical examination, a thorough review of full patient medical history, and neurological testing.
The diagnostic criteria for ALS involve the presence of both upper and lower motor neuron signs in a patient. This can include muscle weakness, muscle wasting, spasticity, and hyperreflexia. The presence of these signs can be confirmed through a thorough physical examination by a neurologist.
Diagnostic testing often includes an electromyogram (EMG) (which tests for muscle activity), a CT scan or MRI (Magnetic Resonance Imaging), and extensive blood work. If necessary, a medical team will perform muscle and/or nerve biopsies to confirm a diagnosis.
Neuroimaging can also be used to support an ALS diagnosis. Magnetic resonance imaging (MRI) of the brain and spinal cord can help rule out other conditions that can mimic ALS, such as spinal cord compression or multiple sclerosis.
However, MRI findings in ALS are often normal in the early stages of the disease.
These tests work to rule out other potential causes of the symptoms.
Currently, there is no specific clinical test for ALS. The lack of a conclusive test can lead to a diagnosis of probable or possible ALS until further identifying symptoms occur. This can be frustrating for patients and their loved ones.
Receiving a confirmed diagnosis of ALS—especially after a long, arduous test process—can be overwhelming for the individual and their family. Anger, denial, and fear are all common reactions. It’s important to talk with your physician and a support group about your personal experience.
Everyone’s journey is different, so it is important to get information and support that is specific to your needs and preferences.
4. Treatment Options

Although there is no cure, there are treatment options for symptoms of ALS. Individuals who have received a medical diagnosis of ALS have, on average, two to five years from the time of the diagnosis.
The progression of ALS varies with each individual. Regular advances in research and healthcare are supporting longer lifespans and more productive lives for a lot of patients.
More than half of those affected live at least three years after diagnosis. Twenty percent will live five years or more, and almost ten percent will live over ten years.
ALS Medications
Riluzole, an oral medication that modestly slows the progression of ALS in some people, is one treatment option for patients with ALS. The exact mechanism for riluzole is unknown; however, researchers believe that it blocks the release of glutamate from nerve cells. Glutamate is a neurotransmitter that is used to relay messages between nerve cells. It is believed that there is an excess of glutamate in individuals with ALS, and this build up damages nerve cells and affects the way they communicate.
When riluzole blocks the release of glutamate from nerve cells, it reduces the build-up of glutamate. This, in turn, reduces the rate of glutamate-induced deterioration and slows the progression of symptoms. For decades, riluzole was the only FDA-approved drug.
The Federal Drug Administration (FDA) recently approved Radicava™ (edaravone), the first new treatment specifically for ALS in 22 years. It is administered by IV infusion for 60 minutes, and it is an ongoing treatment plan. Private duty nurses can manage IV medication administration, wherever the patient is most comfortable. Nurses can also lead teaching visits and administer medication for patients with ALS.
At this time, the mechanism by which Radicava works is unknown. Both Radicava and riluzole do not claim to cure ALS or restore any physical functions that have been lost. Both treatment plans work to slow the progression of the disease.
Radicava has provided an extensive educational webinar on their clinical studies and results, safety information, and more. The video is available to watch on their website, at www.Radicava.com/patient/educational-webinar.
ALS Therapies
As ALS progresses, individuals may require supportive equipment to help with mobility, feeding, and breathing.
Wheelchairs and mobility aids can help individuals maintain independence and mobility. Feeding tubes may be necessary to ensure adequate nutrition, while a tracheostomy may be necessary to help with breathing difficulties.
People with ALS should continue their daily activities, stopping before they get tired.
If you have ALS, set your own limits and plan how you want to use your energy. Physicians may recommend exercises to strengthen affected or unaffected areas; the exercises should not be tiring or strenuous.
The goal is to prevent joint stiffness and muscle contracture and to maintain mobility.
A team of healthcare specialists best suited to the patient’s changing needs can support their goal of a longer lifespan and more productive life. Professionals and treatments can include:
- People with ALS need to be aware that they are receiving enough nutrients, calories, and fluids in order to maintain a normal weight. A registered dietitian or nutritionist can help make sure that a person is receiving enough food and can create solutions as problems with swallowing occur. At some point, the patient may want to consider a feeding tube, referred to as a PEG, which is surgically inserted into the stomach.
- Physical and occupational therapists can identify devices that maximize independence as changes in mobility occur. Devices such as braces, canes, and walkers can help someone with ALS maintain mobility for as long as possible. As the disease progresses, wheelchairs, lifts, and other special equipment can ensure the person receives appropriate care and stays connected with their world.
- A speech therapist can help an individual maintain their speaking ability for as long as possible. As speech abilities deteriorate, people with ALS turn to assistive communicative devices to communicate; this is possible even into the latest stages of ALS. These devices range from simple boards to complex electronic devices and computer applications.
- As the disease progresses, throat muscles weaken and breathing becomes more difficult. This may result in sleeping problems, fatigue, or headaches.
- One of the most commonly used non-invasive options is a BiPAP (biphasic positive airway pressure), which is connected to a mask that is worn over his or her nose at night. It increases the flow of air into and out of the lungs. A private duty nurse can assist by monitoring the BiPAP machine or providing teaching visits.
- When a person requires more help breathing, a permanent ventilator can be used. A ventilator does the person’s breathing for them and is placed via a tracheotomy, where a plastic or metal tube is inserted into the trachea or windpipe. After a tracheotomy, a private nurse can clean wounds and monitor the surgical site to prevent post-surgical infections and reduce the risk of hospital readmission. Most patients use a ventilator 24 hours a day.
Along the ALS journey, patients and their families will need to make tough decisions. Breathing decisions are among some of the most difficult to make. Always be sure to make your physician aware of your preferences regarding all decision-making.
5. Resources
One of the very best steps you can take to assist a loved one with ALS is to hire an in-home nurse.
Nurses are exceptional at providing care, helping with everyday tasks, and ensuring the quality of life for your loved one suffering from ALS.
NurseRegistry is your best private nurse resource if you live in California or Florida. We offer industry-leading flexibility, including 24/7 care if your loved one requires around-the-clock care. We have over 500 professional, licensed, and empathetic nurses on hand and will match you with one that perfectly connects with your loved one – within 48 hours.
Click below to learn more about how NurseRegistry’s private nursing care can help today.

Additional Resources
If you or a loved one has been affected by ALS, these resources are a good starting point to learn more about the disease and develop your support network.
The ALS Association has a mission to discover treatments and a cure for ALS, and to serve, advocate for, and empower people affected by ALS to live their lives to the fullest. The website offers information on the latest research, reading materials, products and services in your local area, and opportunities to raise awareness. Find your local chapter here.
The International Alliance of ALS/MND Associations provides an international community to share resources, advance awareness, and provide support.
The ALS Center at UCSF provides advanced care options. They are focused on the development and application of new treatments.
The Neuromuscular Program at Stanford Health Care provides comprehensive diagnostic evaluation and care for ALS. Open clinical trials for ALS patients can be found here.
Disclaimer: The information provided in this article is not intended to diagnose health problems or take the place of professional medical advice or care you receive from a health care provider. An ALS diagnosis must be confirmed with a licensed medical professional. Always consult your health care provider about symptoms, health problems, medications, and treatments.
Frequently Asked Questions about ALS
What are the initial symptoms of ALS?
The initial symptoms of ALS include muscle weakness, stiffness, and cramping, which can occur in any part of the body. Other symptoms may include difficulty with speaking, swallowing, and breathing. These symptoms may be subtle at first and may be mistaken for other conditions, making early diagnosis difficult.
How is ALS diagnosed?
ALS is typically diagnosed through a combination of physical examination, neurological testing, and diagnostic tests such as electromyography (EMG) and nerve conduction studies. Blood and urine tests may also be performed to rule out other conditions that may mimic ALS symptoms.
What treatment options are available for ALS?
There is currently no cure for ALS, but there are several treatment options available that can help manage symptoms and slow the progression of the disease. These may include medications, physical therapy, speech therapy, and respiratory therapy. In some cases, surgery may be recommended to help manage symptoms.
Are there ways to prevent the onset of ALS?
There is currently no known way to prevent the onset of ALS. However, some lifestyle factors, such as smoking and exposure to certain environmental toxins, may increase the risk of developing the disease.
What is ALS caused by?
The exact cause of ALS is not yet fully understood, but it is thought to be a combination of genetic (family history) and environmental factors. In some cases, the disease may be inherited, while in others, it may occur spontaneously.
What is the typical life expectancy following an ALS diagnosis?
The life expectancy of a person with ALS varies depending on the severity of the disease and other individual factors. On average, people with ALS live 2-5 years after diagnosis, although some may live longer.
How does ALS typically progress in patients?
ALS typically progresses over time, with symptoms becoming more severe and widespread. As the disease progresses, muscle weakness and paralysis may spread to other parts of the body, eventually leading to respiratory failure.
Is ALS a Fatal Disease?
Yes, ALS is a fatal disease. While there is no cure for the disease, treatment can help manage symptoms and improve the quality of life for those living with ALS.



